H3K27me3 summit processing
Renee Matthews
2026-01-23
Last updated: 2026-01-23
Checks: 7 0
Knit directory: DXR_continue/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20250701) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 13ea8c2. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/Bed_exports/
Ignored: data/Cormotif_data/
Ignored: data/DER_data/
Ignored: data/Other_paper_data/
Ignored: data/RDS_files/
Ignored: data/TE_annotation/
Ignored: data/alignment_summary.txt
Ignored: data/all_peak_final_dataframe.txt
Ignored: data/cell_line_info_.tsv
Ignored: data/full_summary_QC_metrics.txt
Ignored: data/motif_lists/
Ignored: data/number_frag_peaks_summary.txt
Untracked files:
Untracked: H3K27ac_all_regions_test.bed
Untracked: H3K27ac_consensus_clusters_test.bed
Untracked: analysis/GREAT_H3K27ac.Rmd
Untracked: analysis/H3K27ac_ChromHMM_FC.Rmd
Untracked: analysis/H3K27ac_cisRE.Rmd
Untracked: analysis/H3K27me3_TE_overlap.Rmd
Untracked: analysis/Top2a_Top2b_expression.Rmd
Untracked: analysis/chromHMM.Rmd
Untracked: analysis/maps_and_plots.Rmd
Untracked: analysis/proteomics.Rmd
Untracked: code/ZNF_genes_bedfile_creation.R
Untracked: other_analysis/
Unstaged changes:
Modified: analysis/H3K27_TE_overlap_extend.Rmd
Modified: analysis/H3K27ac_RNA_integration.Rmd
Modified: analysis/H3K27ac_TF_motifs.Rmd
Modified: analysis/final_analysis.Rmd
Modified: analysis/repeatmasker_data.Rmd
Modified: analysis/summit_files_processing.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown
(analysis/H3K27me3_summit_processing.Rmd) and HTML
(docs/H3K27me3_summit_processing.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 13ea8c2 | reneeisnowhere | 2026-01-23 | first commit |
library(tidyverse)
library(GenomicRanges)
library(plyranges)
library(genomation)
library(readr)
library(rtracklayer)
library(stringr)
library(BiocParallel)
library(parallel)
library(future.apply)sampleinfo <- read_delim("data/sample_info.tsv", delim = "\t")
##Path to histone summit files
H3K27me3_dir <- "C:/Users/renee/Other_projects_data/DXR_data/final_data/summit_files/H3K27me3"
##pull all histone files together
H3K27me3_summit_files <- list.files(
path = H3K27me3_dir,
pattern = "\\.bed$",
recursive = TRUE,
full.names = TRUE
)
length(H3K27me3_summit_files)[1] 29# head(H3K27me3_summit_files)peakAnnoList_H3K27me3 <- readRDS("data/motif_lists/H3K27me3_annotated_peaks.RDS")
H3K27me3_sets_gr <- lapply(peakAnnoList_H3K27me3, function(df) {
as_granges(df)
})
read_summit <- function(file){
peaks <- read.table(file,header = FALSE)
colnames(peaks) <- c("chr","start","end","name","score")
GRanges(
seqnames = peaks$chr,
ranges = IRanges(start=peaks$start, end = peaks$start),
score=peaks$score,
file=basename(file),
Library_ID = stringr::str_remove(basename(file), "_FINAL_summits\\.bed$")
)
}
all_H3K27me3_summits_list<- lapply(H3K27me3_summit_files, read_summit)
all_H3K27me3_summits_gr <- do.call(c, all_H3K27me3_summits_list) # combine into one GRanges object
H3K27me3_lookup <- imap_dfr(peakAnnoList_H3K27me3[1:3], ~
tibble(Peakid = .x@anno$Peakid, cluster = .y)
)
###Adding in sampleinfo dataframe
meta <- as.data.frame(mcols(all_H3K27me3_summits_gr))
meta2 <- meta %>%
left_join(., sampleinfo, by=c("Library_ID"="Library ID"))
mcols(all_H3K27me3_summits_gr) <- meta2
mcols(all_H3K27me3_summits_gr)$group <-
paste(all_H3K27me3_summits_gr$Treatment,
all_H3K27me3_summits_gr$Timepoint,
sep = "_")
ROIs <- H3K27me3_sets_gr$all_H3K27me3# -------------------------
# Step 1: Reduce within groups (parallel, Windows-safe)
# -------------------------
get_highest_per_group_parallel <- function(summits_gr, group_col = "group",
score_col = "score", min_gap_within = 100,
workers = 2) {
# Split GRanges by group to reduce memory per worker
group_list <- split(summits_gr, mcols(summits_gr)[[group_col]])
plan(multisession, workers = workers) # Windows-compatible
results <- future_lapply(group_list, function(gr_sub) {
if (length(gr_sub) == 0) return(GRanges())
red <- GenomicRanges::reduce(gr_sub, min.gapwidth = min_gap_within, ignore.strand = TRUE, with.revmap = TRUE)
revmap <- mcols(red)$revmap
idx <- unlist(lapply(revmap, function(x) {
scores <- mcols(gr_sub)[[score_col]][x]
x[which.max(scores)]
}))
gr_sub[idx]
}, future.seed = TRUE)
do.call(c, results)
}
# -------------------------
# Step 2: Reduce across groups (parallel, Windows-safe)
# -------------------------
get_consensus_summits_parallel <- function(highest_per_group_gr, score_col = "score",
min_gap_across = 400, workers = 2) {
if (length(highest_per_group_gr) == 0) return(GRanges())
# Split by chromosome to reduce memory per worker
chr_list <- split(highest_per_group_gr, seqnames(highest_per_group_gr))
plan(multisession, workers = workers)
results <- future_lapply(chr_list, function(gr_sub) {
red <- GenomicRanges::reduce(gr_sub, min.gapwidth = min_gap_across, ignore.strand = TRUE, with.revmap = TRUE)
revmap <- mcols(red)$revmap
idx <- unlist(lapply(revmap, function(x) {
scores <- mcols(gr_sub)[[score_col]][x]
x[which.max(scores)]
}))
gr_sub[idx]
}, future.seed = TRUE)
do.call(c, results)
}
####### step 3 #####################3
assign_best_summit_to_ROI_parallel <- function(consensus_gr,
ROIs_gr,
max_dist = 500,
workers = 2) {
# Split by chromosome
roi_list <- split(ROIs_gr, seqnames(ROIs_gr))
cons_list <- split(consensus_gr, seqnames(consensus_gr))
plan(multisession, workers = workers)
results <- future_lapply(intersect(names(roi_list), names(cons_list)),
function(chr) {
rois_chr <- roi_list[[chr]]
cons_chr <- cons_list[[chr]]
if (length(rois_chr) == 0) return(tibble())
# -----------------------------
# ROI metadata (ground truth)
# -----------------------------
roi_meta <- tibble(
Peakid = rois_chr$Peakid,
roi_seqname = as.character(seqnames(rois_chr)),
roi_start = start(rois_chr),
roi_end = end(rois_chr)
)
# -----------------------------
# 1) Exact overlaps
# -----------------------------
ov <- findOverlaps(rois_chr, cons_chr)
assigned_df <- if (length(ov) > 0) {
tibble(
Peakid = rois_chr$Peakid[queryHits(ov)],
summit_pos = start(cons_chr)[subjectHits(ov)],
summit_score = mcols(cons_chr)$score[subjectHits(ov)]
) %>%
group_by(Peakid) %>%
slice_max(summit_score, with_ties = FALSE) %>%
ungroup()
} else {
tibble()
}
# -----------------------------
# 2) Nearest fallback
# -----------------------------
unassigned_peaks <- setdiff(roi_meta$Peakid, assigned_df$Peakid)
if (length(unassigned_peaks) > 0 && length(cons_chr) > 0) {
roi_unassigned <- rois_chr[rois_chr$Peakid %in% unassigned_peaks]
dn <- distanceToNearest(roi_unassigned, cons_chr)
dn_df <- tibble(
Peakid = roi_unassigned$Peakid[queryHits(dn)],
summit_pos = start(cons_chr)[subjectHits(dn)],
summit_score = mcols(cons_chr)$score[subjectHits(dn)],
distance = mcols(dn)$distance
) %>%
filter(distance <= max_dist) %>%
group_by(Peakid) %>%
slice_max(summit_score, with_ties = FALSE) %>%
ungroup()
assigned_df <- bind_rows(assigned_df, dn_df)
}
# -----------------------------
# 3) Merge + derived metrics
# -----------------------------
out_df <- left_join(roi_meta, assigned_df, by = "Peakid") %>%
mutate(
roi_center = roi_start + (roi_end - roi_start) / 2,
dist_center = ifelse(is.na(summit_pos),
NA_real_,
summit_pos - roi_center),
rel_pos = ifelse(is.na(summit_pos),
NA_real_,
(summit_pos - roi_start) / (roi_end - roi_start))
)
out_df
}, future.seed = TRUE)
final_df <- bind_rows(results)
# -----------------------------
# 4) GRanges of assigned summits
# -----------------------------
assigned_rows <- final_df %>% filter(!is.na(summit_pos))
assigned_gr <- if (nrow(assigned_rows) > 0) {
GRanges(
seqnames = assigned_rows$roi_seqname,
ranges = IRanges(
start = assigned_rows$summit_pos,
end = assigned_rows$summit_pos
),
Peakid = assigned_rows$Peakid,
score = assigned_rows$summit_score,
dist_center = assigned_rows$dist_center,
rel_pos = assigned_rows$rel_pos
)
} else {
GRanges()
}
list(df = final_df, gr = assigned_gr)
}Evaluation of gap width in summits
Reduction plots
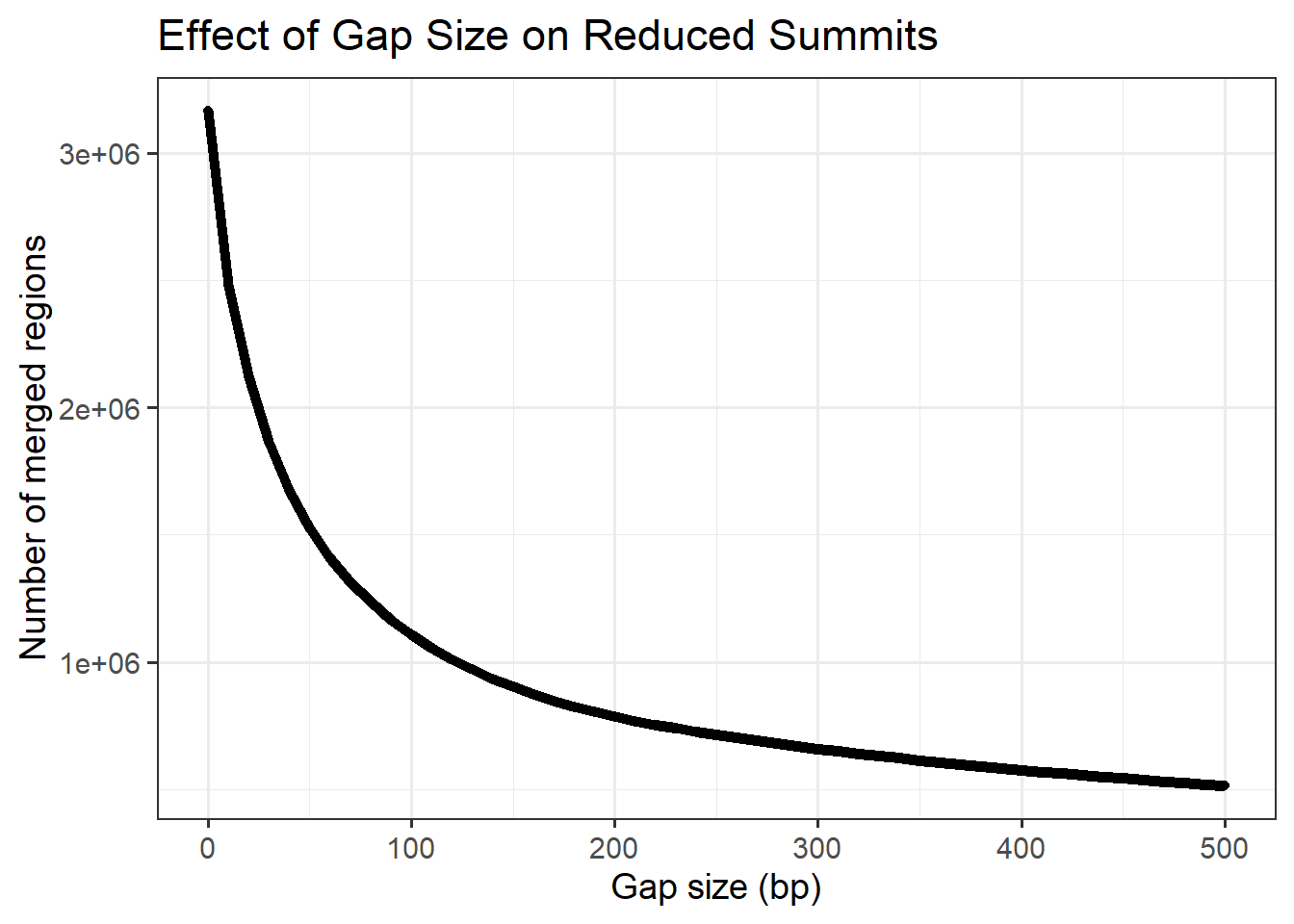
Plotting effect of reduction bp number on total number of clusters
gap_sizes <- seq(0, 500, by = 10)
# Function to apply reduce for each gap and return counts
results <- lapply(gap_sizes, function(g) {
reduced <- GenomicRanges::reduce(all_H3K27me3_summits_gr, min.gapwidth = g)
data.frame(gap = g, n_regions = length(reduced))
})
# Combine results
min_gap_summary <- bind_rows(results)
ggplot(min_gap_summary, aes(x = gap, y= n_regions))+
geom_line(linewidth=2)+
geom_point()+
theme_bw(base_size=14)+
labs(
title = "Effect of Gap Size on Reduced Summits",
x = "Gap size (bp)",
y = "Number of merged regions"
)
# Function to pick highest summit per cluster from a reduced GRanges list
get_highest_per_group <- function(reduced_groups, orig_summits_gr, group_col = "group") {
groups <- names(reduced_groups)
highest_per_group <- vector("list", length(reduced_groups))
names(highest_per_group) <- groups
for(i in seq_along(reduced_groups)) {
gr <- reduced_groups[[i]]
# original summits for this group
orig <- orig_summits_gr[mcols(orig_summits_gr)[[group_col]] == groups[i]]
scores <- orig$score
# revmap is a CompressedIntegerList
revmap <- mcols(gr)$revmap
# skip if revmap is NULL
if(is.null(revmap)) next
# unlist all indices once
all_idx <- unlist(revmap, use.names = FALSE)
# repeat cluster index for each element in revmap
cluster_idx <- rep(seq_along(revmap), times = elementNROWS(revmap))
# scores for all indices
all_scores <- scores[all_idx]
# For each cluster, pick the index of the max score
max_idx_per_cluster <- tapply(seq_along(all_scores), cluster_idx, function(ii) {
ii[which.max(all_scores[ii])]
})
# Convert back to original indices
orig_idx <- all_idx[unlist(max_idx_per_cluster)]
# subset original GRanges
highest_per_group[[i]] <- orig[orig_idx]
}
# Flatten any nested GRangesList
flatten_gr <- function(x) {
if (inherits(x, "GRanges")) return(x)
if (inherits(x, "GRangesList")) return(unlist(x, use.names = FALSE))
if (is.list(x)) return(do.call(c, lapply(x, flatten_gr)))
stop("Unexpected object type")
}
highest_summits_gr <- flatten_gr(highest_per_group)
# Return as long GRanges with group column
highest_summits_df <- bind_rows(
lapply(names(highest_per_group), function(gr_name) {
as.data.frame(highest_per_group[[gr_name]]) %>%
mutate(group = gr_name)
})
)
highest_summits_long_gr <- highest_summits_df %>% GRanges()
return(highest_summits_long_gr)
}###now splitting into grouped granges
gr_by_group <- split(all_H3K27me3_summits_gr,
all_H3K27me3_summits_gr$group)
# gr_by_group <- as(gr_by_group, "CompressedGRangesList")
### now reducing within some width by 100 bp with revmap
groups <- unique(all_H3K27me3_summits_gr$group)
reduced_groups <- lapply(groups, function(g) {
gr_sub <- all_H3K27me3_summits_gr[all_H3K27me3_summits_gr$group == g]
GenomicRanges::reduce(gr_sub, min.gapwidth = 100, ignore.strand = TRUE, with.revmap = TRUE)
})
reduced_groups_200 <- lapply(groups, function(g) {
gr_sub <- all_H3K27me3_summits_gr[all_H3K27me3_summits_gr$group == g]
GenomicRanges::reduce(gr_sub, min.gapwidth = 200, ignore.strand = TRUE, with.revmap = TRUE)
})
reduced_groups_300 <- lapply(groups, function(g) {
gr_sub <- all_H3K27me3_summits_gr[all_H3K27me3_summits_gr$group == g]
GenomicRanges::reduce(gr_sub, min.gapwidth = 300, ignore.strand = TRUE, with.revmap = TRUE)
})
reduced_groups_400 <- lapply(groups, function(g) {
gr_sub <- all_H3K27me3_summits_gr[all_H3K27me3_summits_gr$group == g]
GenomicRanges::reduce(gr_sub, min.gapwidth = 400, ignore.strand = TRUE, with.revmap = TRUE)
})
names(reduced_groups) <- groups
names(reduced_groups_200) <- groups
names(reduced_groups_300) <- groups
names(reduced_groups_400) <- groupsreduced_sets <- list(
"100bp" = reduced_groups,
"200bp" = reduced_groups_200,
"300bp" = reduced_groups_300,
"400bp" = reduced_groups_400
)
# highest_summits_all <- lapply(reduced_sets, get_highest_per_group, orig_summits_gr = all_H3K27me3_summits_gr)
# saveRDS(highest_summits_all,"data/RDS_files/H3K27me3_highest_summits_all.RDS")
highest_summits_all <- readRDS("data/RDS_files/H3K27me3_highest_summits_all.RDS")Looking at number of summits across groups as a function of min.gap number
##Compute counts per group for each reduced set
summit_counts_group <- lapply(names(highest_summits_all), function(gap_name) {
gr <- highest_summits_all[[gap_name]]
# make sure group column exists
if(!"group" %in% colnames(mcols(gr))) stop("GRanges must have 'group' column")
df <- as.data.frame(gr) %>%
count(group, name = "n_summits") %>%
mutate(gap = gap_name)
return(df)
}) %>% bind_rows()
ggplot(summit_counts_group, aes(x = gap, y = n_summits, group = group, color = group)) +
geom_line(size = 1.2) +
geom_point(size = 3) +
theme_classic(base_size = 14) +
labs(
title = "Effect of min-gap width on number of highest summits\nper group; H3K27me3",
x = "Min-gap width (bp)",
y = "Number of highest summits",
color = "Group"
) +
theme(axis.text.x = element_text(angle = 45, hjust = 1))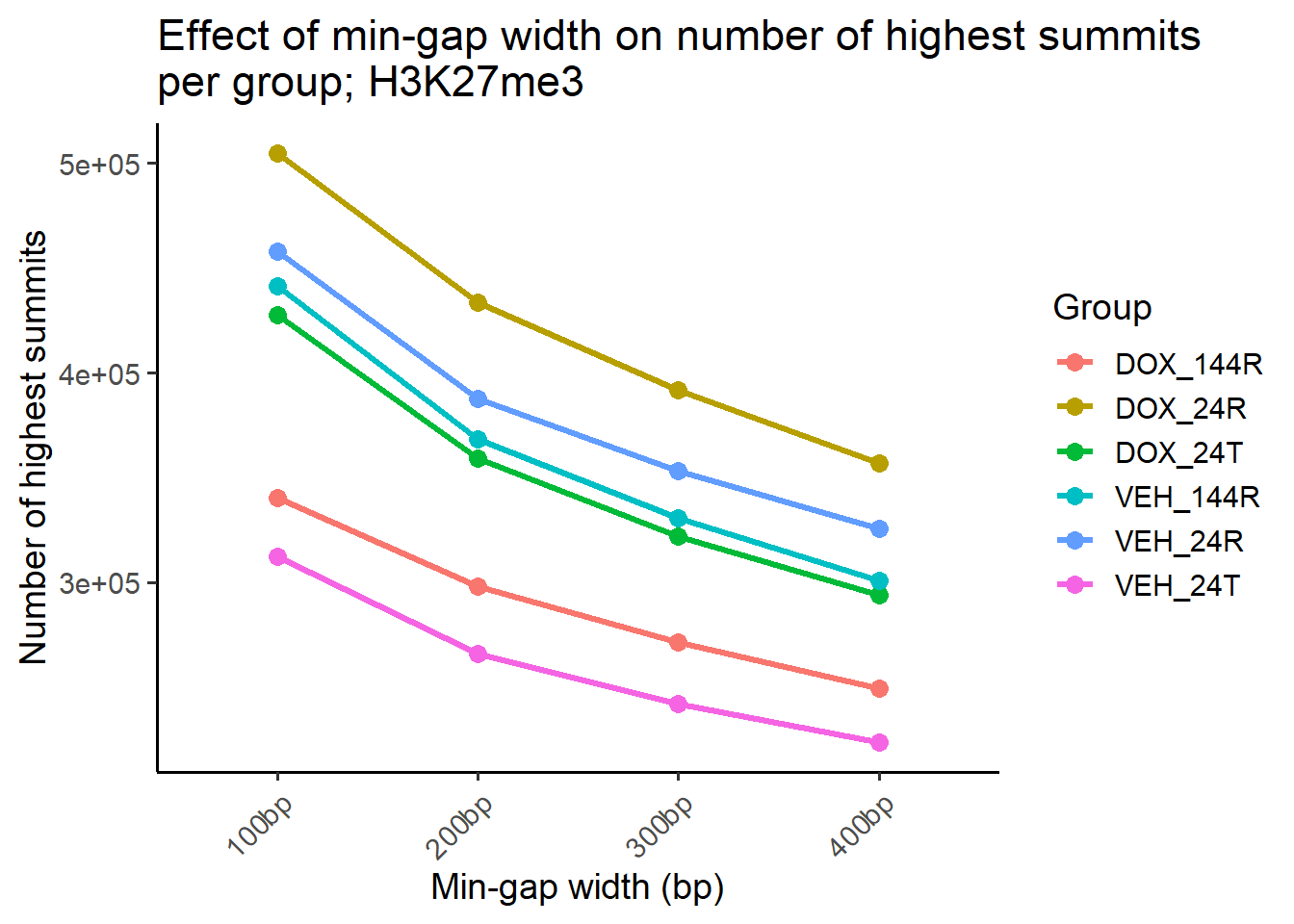 ### Summit average per ROI What is the average number of summits per ROI
as a function of min.gapwidth
### Summit average per ROI What is the average number of summits per ROI
as a function of min.gapwidth
double_reduce_summits <- function(summits_gr, ROIs, groups,
min_gap_within_seq = c(100,200,300),
min_gap_across_seq = c(100,200,300),
BPPARAM = MulticoreParam(4)) {
# Pre-split summits by group to avoid repeated subsetting
summits_by_group <- split(summits_gr, summits_gr$group)
# Create all gap combinations
gap_combos <- expand.grid(min_gap_within = min_gap_within_seq,
min_gap_across = min_gap_across_seq,
stringsAsFactors = FALSE)
# Apply in parallel for each combination
results <- bplapply(seq_len(nrow(gap_combos)), function(i) {
g1 <- gap_combos$min_gap_within[i]
g2 <- gap_combos$min_gap_across[i]
# -----------------
# Step 1: Reduce within group
# -----------------
highest_per_group <- lapply(groups, function(grp) {
gr_sub <- summits_by_group[[grp]]
if(length(gr_sub) == 0) return(NULL)
# reduce within group with revmap
red <- GenomicRanges::reduce(gr_sub, min.gapwidth = g1, ignore.strand = TRUE, with.revmap = TRUE)
revmap <- mcols(red)$revmap
if(length(revmap) == 0) return(NULL)
# pick highest score per cluster
cluster_idx <- rep(seq_along(revmap), times = elementNROWS(revmap))
all_idx <- unlist(revmap, use.names = FALSE)
all_scores <- gr_sub$score[all_idx]
max_idx_per_cluster <- tapply(seq_along(all_scores), cluster_idx, function(ii) {
ii[which.max(all_scores[ii])]
})
gr_sub[all_idx[unlist(max_idx_per_cluster)]]
})
highest_per_group <- highest_per_group[!sapply(highest_per_group, is.null)]
# -----------------
# Step 2: Merge across groups
# -----------------
if(length(highest_per_group) == 0) return(NULL)
all_highest <- do.call(c, highest_per_group)
consensus <- GenomicRanges::reduce(all_highest, min.gapwidth = g2, ignore.strand = TRUE)
# -----------------
# Step 3: Count summits per ROI (vectorized)
# -----------------
hits <- findOverlaps(ROIs, consensus)
counts <- as.data.frame(table(queryHits(hits)))
colnames(counts) <- c("ROI_idx", "n_summits")
counts$ROI_idx <- as.integer(as.character(counts$ROI_idx))
counts$min_gap_within <- g1
counts$min_gap_across <- g2
counts
}, BPPARAM = BPPARAM)
# Combine results
results_df <- bind_rows(results)
return(results_df)
}# BPPARAM <- SnowParam(workers = 4, type = "SOCK")
# register(BPPARAM)
#
# heatmap_data <- double_reduce_summits(
# summits_gr = all_H3K27me3_summits_gr,
# ROIs = H3K27me3_sets_gr$all_H3K27me3,
# groups = unique(all_H3K27me3_summits_gr$group),
# min_gap_within_seq = c(100,200,300,400),
# min_gap_across_seq = c(100,200,300,400),
# BPPARAM = BPPARAM
# )
# saveRDS(heatmap_data,"data/RDS_files/H3K27me_heatmap_data.RDS")
heatmap_data <- readRDS("data/RDS_files/H3K27me_heatmap_data.RDS")
heatmap_avg <- heatmap_data %>%
group_by(min_gap_within, min_gap_across) %>%
summarise(mean_summits = mean(n_summits, na.rm = TRUE), .groups = "drop")Exploring differences in min gaps in step 1 and step 2 reductions
ggplot(heatmap_avg, aes(x = factor(min_gap_within),
y = factor(min_gap_across),
fill = mean_summits)) +
geom_tile(color = "white") +
geom_text(aes(label = round(mean_summits, 1)), color = "black", size = 4) +
scale_fill_viridis_c(option = "plasma") +
labs(
x = "Within-group min-gap (bp)",
y = "Across-group min-gap (bp)",
fill = "Mean # summits per ROI",
title = "Effect of min-gap width on summits per ROI\nH3K27me3"
) +
theme_minimal(base_size = 14) +
theme(axis.text.x = element_text(angle = 45, hjust = 1))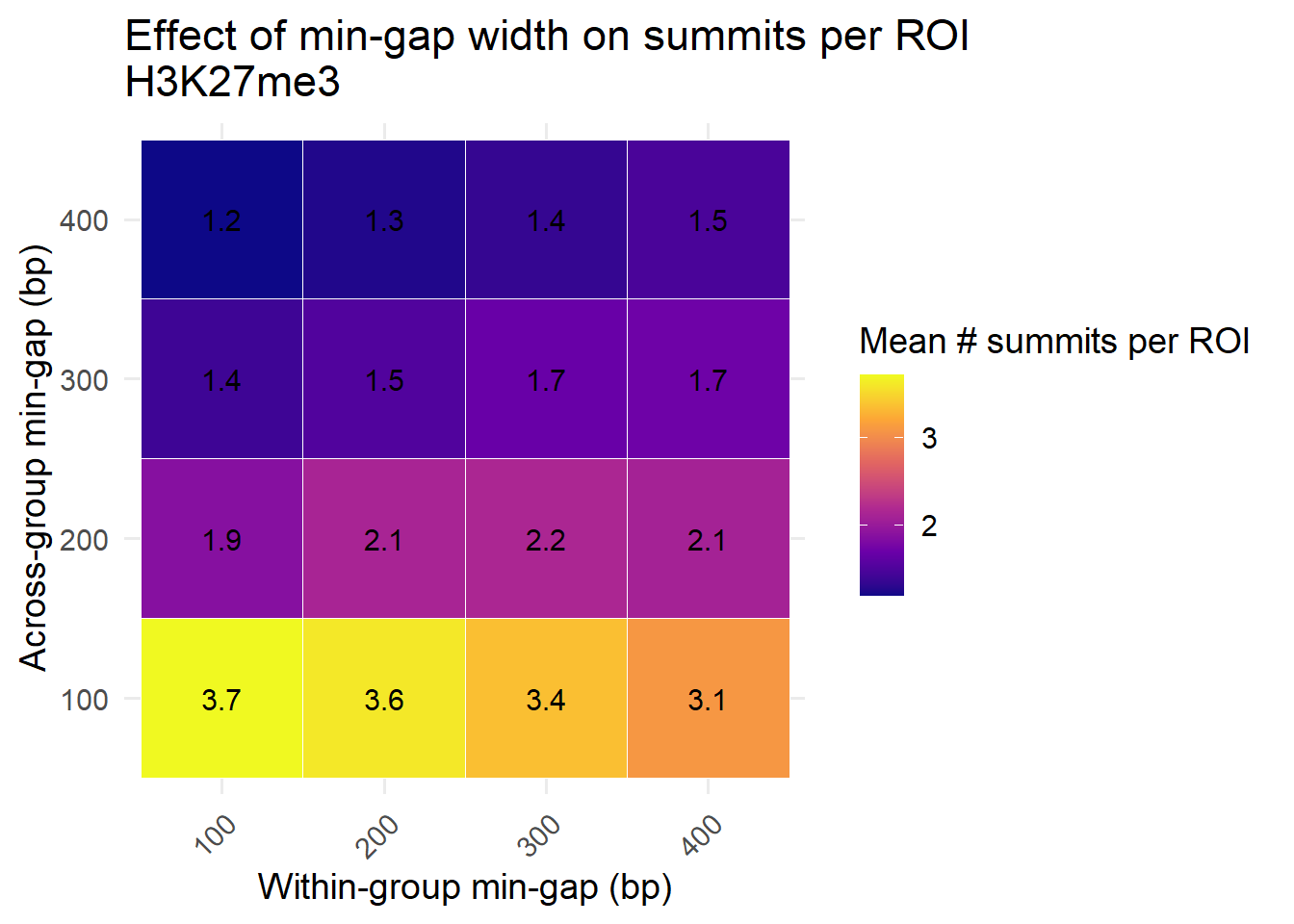
# options(future.globals.maxSize = 10 * 1024^3)
# workers <- parallel::detectCores() - 1
# ### Step 1
# temp_highest_per_group <- get_highest_per_group_parallel(all_H3K27me3_summits_gr,
# group_col = "group",
# score_col = "score",
# min_gap_within = 100,
# workers = workers)
# saveRDS(temp_highest_per_group,"data/RDS_files/H3K27me3_temp_highest_per_group.RDS")
temp_highest_per_group <- readRDS("data/RDS_files/H3K27me3_temp_highest_per_group.RDS")
# Concatenate into one GRanges (Step 1.2)
flat_highest_gr <- (c(temp_highest_per_group$DOX_144R, temp_highest_per_group$DOX_24R,temp_highest_per_group$DOX_24T,temp_highest_per_group$VEH_144R,temp_highest_per_group$VEH_24R,temp_highest_per_group$VEH_24T))
### STep 2
# consensus_summits <- get_consensus_summits_parallel(
# flat_highest_gr,
# score_col = "score",
# min_gap_across = 400,
# workers = workers
# )
####Concatenate into one GRanges
# consensus_summits_gr <- (c(consensus_summits$chr1,consensus_summits$chr2,
# consensus_summits$chr3,consensus_summits$chr4,
# consensus_summits$chr5,consensus_summits$chr6,
# consensus_summits$chr7,consensus_summits$chr8,
# consensus_summits$chr9,consensus_summits$chr10,
# consensus_summits$chr11,consensus_summits$chr12,
# consensus_summits$chr13,consensus_summits$chr14,
# consensus_summits$chr15,consensus_summits$chr16,
# consensus_summits$chr17,consensus_summits$chr18,
# consensus_summits$chr19,consensus_summits$chr20,
# consensus_summits$chr21,consensus_summits$chr22))
# saveRDS(consensus_summits_gr,"data/RDS_files/H3K27me3_consensus_summits_gr.RDS")
consensus_summits_gr <- readRDS("data/RDS_files/H3K27me3_consensus_summits_gr.RDS")
# final_data <- assign_best_summit_to_ROI_parallel(consensus_summits_gr, ROIs,max_dist = 500,workers = workers)
# saveRDS(final_data,"data/RDS_files/H3K27me3_final_data.RDS")
final_data <- readRDS("data/RDS_files/H3K27me3_final_data.RDS")
# data.frame with one row per ROI (assigned or NA)
final_df <- final_data$df
# GRanges of ROIs that received an assigned summit (with metadata)
assigned_summits_gr <- final_data$gr
# Quick checks
n_missing <- sum(is.na(final_df$summit_pos))
cat("ROIs lacking any summit within max_dist:", n_missing, "\n")ROIs lacking any summit within max_dist: 8383 na_rois <- final_df %>% filter(is.na(summit_pos))
na_rois_gr <- GRanges(
seqnames = na_rois$roi_seqname,
ranges = IRanges(start = na_rois$roi_start, end = na_rois$roi_end),
Peakid = na_rois$Peakid
)
ov <- findOverlaps(na_rois_gr, flat_highest_gr)
overlap_df <- tibble(
roi_idx = queryHits(ov),
summit_idx = subjectHits(ov),
summit_pos = start(all_H3K27me3_summits_gr)[subjectHits(ov)],
summit_score = mcols(all_H3K27me3_summits_gr)$score[subjectHits(ov)]
)
best_summits <- overlap_df %>%
group_by(roi_idx) %>%
slice_max(summit_score, with_ties = FALSE) %>%
ungroup()
assigned_df <- tibble(
roi_idx = seq_along(na_rois_gr),
Peakid = na_rois_gr$Peakid,
roi_seqname = as.character(seqnames(na_rois_gr)),
roi_start = start(na_rois_gr),
roi_end = end(na_rois_gr)
) %>%
left_join(best_summits, by = "roi_idx")
assigned_df_calc <- assigned_df %>% mutate(
dist_center = summit_pos - (roi_start + (roi_end - roi_start)/2),
rel_pos = (summit_pos - roi_start)/(roi_end - roi_start)
)
complete_summit_df <- final_df %>%
dplyr::select(!distance) %>%
dplyr::filter(!is.na(summit_pos)) %>%
bind_rows(.,assigned_df_calc) %>%
left_join(., H3K27me3_lookup, by = "Peakid") %>%
dplyr::select(!summit_idx) %>%
# dplyr::select(!cons_idx) %>%
group_by(Peakid) %>%
slice_min(order_by = abs(dist_center), n = 1) %>%
distinct
# saveRDS(complete_summit_df,"data/RDS_files/H3K27me3_complete_summit_df.RDS")
only_complete <- complete_summit_df %>% filter(!is.na(summit_pos))
complete_summit_gr <- GRanges(
seqnames=only_complete$roi_seqname,
ranges=IRanges(start= only_complete$summit_pos,
end= only_complete$summit_pos))
# Columns to exclude from metadata (used to define GRanges)
exclude_cols <- c("roi_seqname", "summit_pos")
# Only keep columns that exist in df
meta_cols <- intersect(setdiff(colnames(only_complete), exclude_cols), colnames(only_complete))
# Assign metadata
mcols(complete_summit_gr) <- only_complete[, meta_cols, drop = FALSE]
# saveRDS(complete_summit_gr,"data/RDS_files/H3K27me3_complete_summit_gr.RDS")### adding in cluster membership for export
library(BSgenome.Hsapiens.UCSC.hg38)
genome <- BSgenome.Hsapiens.UCSC.hg38
seqlengths(complete_summit_gr) <- seqlengths(genome)[names(seqlengths(complete_summit_gr))]
SET_1_gr <- complete_summit_gr[
!is.na(mcols(complete_summit_gr)$cluster) &
mcols(complete_summit_gr)$cluster == "Set_1"
]
resize_and_trim <- function(gr, flank = 300, genome = BSgenome.Hsapiens.UCSC.hg38) {
gr <- resize(gr, width = 1 + 2*flank, fix = "center")
seqlengths(gr) <- seqlengths(genome)[seqlevels(gr)]
gr <- trim(gr)
gr <- gr[width(gr) > 0]
return(gr)
}
# H3K27me3_set1_600 <- resize_and_trim(SET_1_gr,flank=300)
H3K27me3_set1_400 <- resize_and_trim(SET_1_gr,flank=200)
# H3K27me3_set1_600<- trim(H3K27me3_set1_600)
# H3K27me3_set1_600 <- H3K27me3_set1_600[width(H3K27me3_set1_600) > 0]
H3K27me3_set1_400 <- resize(SET_1_gr, width = 1 + 200*2, fix = "center")
H3K27me3_set1_400<- trim(H3K27me3_set1_400)
H3K27me3_set1_400 <- H3K27me3_set1_400[width(H3K27me3_set1_400) > 0]
rtracklayer::export(SET_1_gr, "data/Bed_exports/H3K27me3_Set_1_summits.bed")
rtracklayer::export(H3K27me3_set1_600, "data/Bed_exports/H3K27me3_Set_1_600.bed")
rtracklayer::export(H3K27me3_set1_400, "data/Bed_exports/H3K27me3_Set_1_400.bed")
SET_2_gr <- complete_summit_gr[
!is.na(mcols(complete_summit_gr)$cluster) &
mcols(complete_summit_gr)$cluster == "Set_2"]
# H3K27me3_set2_600 <- resize_and_trim(SET_2_gr,flank=300)
H3K27me3_set2_400 <- resize_and_trim(SET_2_gr,flank=400)
# H3K27me3_set2_600 <- resize(SET_2_gr, width = 1 + 300*2, fix = "center")
# H3K27me3_set2_600 <- trim(H3K27me3_set2_600)
# H3K27me3_set2_600 <- H3K27me3_set2_600[width(H3K27me3_set2_600) > 0]
H3K27me3_set2_400 <- resize(SET_2_gr, width = 1 + 200*2, fix = "center")
H3K27me3_set2_400 <- trim(H3K27me3_set2_400)
H3K27me3_set2_400 <- H3K27me3_set2_400[width(H3K27me3_set2_400) > 0]
rtracklayer::export(SET_2_gr, "data/Bed_exports/H3K27me3_Set_2_summits.bed")
# rtracklayer::export(H3K27me3_set2_600, "data/Bed_exports/H3K27me3_Set_2_600.bed")
rtracklayer::export(H3K27me3_set2_400, "data/Bed_exports/H3K27me3_Set_2_400.bed")
#
# SET_3_gr <- complete_summit_gr[
# !is.na(mcols(complete_summit_gr)$cluster) &
# mcols(complete_summit_gr)$cluster == "Set_3"]
#
# H3K27me3_set3_600 <- resize_and_trim(SET_3_gr,flank=300)
# H3K27me3_set3_400 <- resize_and_trim(SET_3_gr,flank=200)
#
# H3K27me3_set3_600 <- resize(SET_3_gr, width = 1 + 300*2, fix = "center")
# H3K27me3_set3_600 <- trim(H3K27me3_set3_600)
# H3K27me3_set3_600 <- H3K27me3_set3_600[width(H3K27me3_set3_600) > 0]
#
# H3K27me3_set3_400 <- resize(SET_3_gr, width = 1 + 200*2, fix = "center")
# H3K27me3_set3_400 <- trim(H3K27me3_set3_400)
# H3K27me3_set3_400 <- H3K27me3_set3_400[width(H3K27me3_set3_400) > 0]
#
# rtracklayer::export(SET_3_gr, "data/Bed_exports/H3K27me3_Set_3_summits.bed")
# rtracklayer::export(H3K27me3_set3_600, "data/Bed_exports/H3K27me3_Set_3_600.bed")
# rtracklayer::export(H3K27me3_set3_400, "data/Bed_exports/H3K27me3_Set_3_400.bed")
rtracklayer::export(complete_summit_gr, "data/Bed_exports/H3K27me3_complete_final_summits.bed")
outdir <- "data/Bed_exports/summit_groups/"
dir.create(outdir, showWarnings = FALSE)
group_gr_list <- split(consensus_summits_gr, consensus_summits_gr$group)
for (nm in names(group_gr_list)) {
outfile <- file.path(outdir, paste0(nm, "_H3K27me3_summits.bed"))
export(group_gr_list[[nm]], outfile, format = "BED")
}
names(group_gr_list)
sessionInfo()R version 4.4.2 (2024-10-31 ucrt)
Platform: x86_64-w64-mingw32/x64
Running under: Windows 11 x64 (build 26200)
Matrix products: default
locale:
[1] LC_COLLATE=English_United States.utf8
[2] LC_CTYPE=English_United States.utf8
[3] LC_MONETARY=English_United States.utf8
[4] LC_NUMERIC=C
[5] LC_TIME=English_United States.utf8
time zone: America/Chicago
tzcode source: internal
attached base packages:
[1] parallel grid stats4 stats graphics grDevices utils
[8] datasets methods base
other attached packages:
[1] ChIPseeker_1.42.1 future.apply_1.20.0 future_1.67.0
[4] BiocParallel_1.40.2 rtracklayer_1.66.0 genomation_1.38.0
[7] plyranges_1.26.0 GenomicRanges_1.58.0 GenomeInfoDb_1.42.3
[10] IRanges_2.40.1 S4Vectors_0.44.0 BiocGenerics_0.52.0
[13] lubridate_1.9.4 forcats_1.0.0 stringr_1.5.1
[16] dplyr_1.1.4 purrr_1.1.0 readr_2.1.5
[19] tidyr_1.3.1 tibble_3.3.0 ggplot2_3.5.2
[22] tidyverse_2.0.0 workflowr_1.7.1
loaded via a namespace (and not attached):
[1] RColorBrewer_1.1-3
[2] rstudioapi_0.17.1
[3] jsonlite_2.0.0
[4] magrittr_2.0.3
[5] ggtangle_0.0.7
[6] GenomicFeatures_1.58.0
[7] farver_2.1.2
[8] rmarkdown_2.29
[9] fs_1.6.6
[10] BiocIO_1.16.0
[11] zlibbioc_1.52.0
[12] vctrs_0.6.5
[13] memoise_2.0.1
[14] Rsamtools_2.22.0
[15] RCurl_1.98-1.17
[16] ggtree_3.14.0
[17] htmltools_0.5.8.1
[18] S4Arrays_1.6.0
[19] TxDb.Hsapiens.UCSC.hg19.knownGene_3.2.2
[20] plotrix_3.8-4
[21] curl_7.0.0
[22] gridGraphics_0.5-1
[23] SparseArray_1.6.2
[24] sass_0.4.10
[25] parallelly_1.45.1
[26] KernSmooth_2.23-26
[27] bslib_0.9.0
[28] plyr_1.8.9
[29] impute_1.80.0
[30] cachem_1.1.0
[31] GenomicAlignments_1.42.0
[32] igraph_2.1.4
[33] whisker_0.4.1
[34] lifecycle_1.0.4
[35] pkgconfig_2.0.3
[36] Matrix_1.7-3
[37] R6_2.6.1
[38] fastmap_1.2.0
[39] GenomeInfoDbData_1.2.13
[40] MatrixGenerics_1.18.1
[41] enrichplot_1.26.6
[42] digest_0.6.37
[43] aplot_0.2.8
[44] colorspace_2.1-1
[45] patchwork_1.3.2
[46] AnnotationDbi_1.68.0
[47] ps_1.9.1
[48] rprojroot_2.1.1
[49] RSQLite_2.4.3
[50] labeling_0.4.3
[51] timechange_0.3.0
[52] httr_1.4.7
[53] abind_1.4-8
[54] compiler_4.4.2
[55] bit64_4.6.0-1
[56] withr_3.0.2
[57] DBI_1.2.3
[58] gplots_3.2.0
[59] R.utils_2.13.0
[60] rappdirs_0.3.3
[61] DelayedArray_0.32.0
[62] rjson_0.2.23
[63] caTools_1.18.3
[64] gtools_3.9.5
[65] tools_4.4.2
[66] ape_5.8-1
[67] httpuv_1.6.16
[68] R.oo_1.27.1
[69] glue_1.8.0
[70] restfulr_0.0.16
[71] callr_3.7.6
[72] nlme_3.1-168
[73] GOSemSim_2.32.0
[74] promises_1.3.3
[75] getPass_0.2-4
[76] gridBase_0.4-7
[77] reshape2_1.4.4
[78] fgsea_1.32.4
[79] generics_0.1.4
[80] gtable_0.3.6
[81] BSgenome_1.74.0
[82] tzdb_0.5.0
[83] R.methodsS3_1.8.2
[84] seqPattern_1.38.0
[85] data.table_1.17.8
[86] hms_1.1.3
[87] XVector_0.46.0
[88] ggrepel_0.9.6
[89] pillar_1.11.0
[90] yulab.utils_0.2.1
[91] vroom_1.6.5
[92] later_1.4.2
[93] splines_4.4.2
[94] treeio_1.30.0
[95] lattice_0.22-7
[96] bit_4.6.0
[97] tidyselect_1.2.1
[98] GO.db_3.20.0
[99] Biostrings_2.74.1
[100] knitr_1.50
[101] git2r_0.36.2
[102] SummarizedExperiment_1.36.0
[103] xfun_0.52
[104] Biobase_2.66.0
[105] matrixStats_1.5.0
[106] stringi_1.8.7
[107] UCSC.utils_1.2.0
[108] lazyeval_0.2.2
[109] boot_1.3-32
[110] ggfun_0.2.0
[111] yaml_2.3.10
[112] evaluate_1.0.5
[113] codetools_0.2-20
[114] qvalue_2.38.0
[115] ggplotify_0.1.2
[116] cli_3.6.5
[117] processx_3.8.6
[118] jquerylib_0.1.4
[119] dichromat_2.0-0.1
[120] Rcpp_1.1.0
[121] globals_0.18.0
[122] png_0.1-8
[123] XML_3.99-0.18
[124] blob_1.2.4
[125] DOSE_4.0.1
[126] bitops_1.0-9
[127] listenv_0.9.1
[128] viridisLite_0.4.2
[129] tidytree_0.4.6
[130] scales_1.4.0
[131] crayon_1.5.3
[132] rlang_1.1.6
[133] fastmatch_1.1-6
[134] cowplot_1.2.0
[135] KEGGREST_1.46.0